
前言
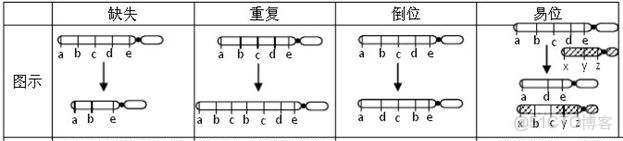
SV(大片段結構變異)指在基因組水平上大片段的insertion,deletion,inversion,translocation,duplication等變異。

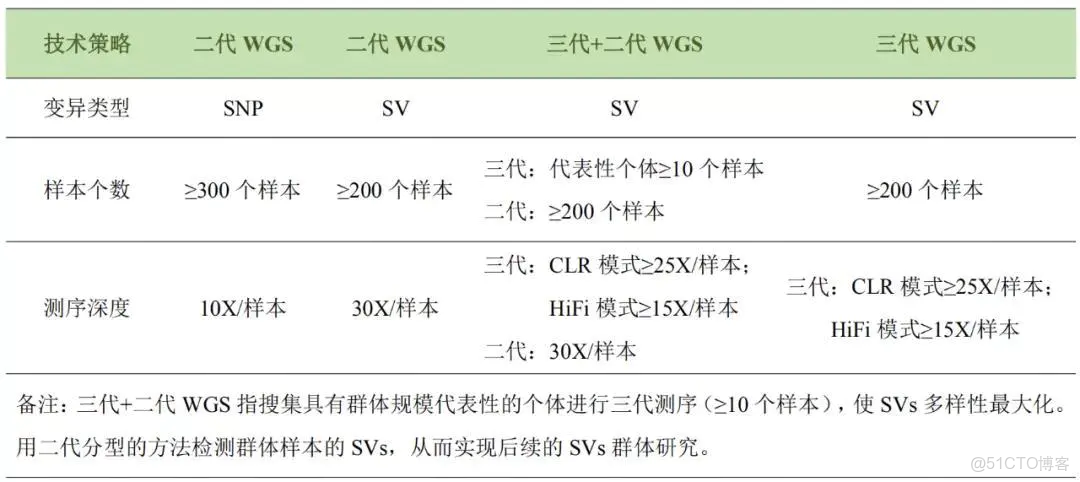
SV檢測分重測序部分和基因組部分,重測序又分二代測序數據和三代測序數據之分。每種分析方法用到的軟件是不一樣的,但結果可能有重疊部分。
針對於基因組,檢測出的SV的準確度主要取決於基因組組裝質量,三代組裝的結果更優於二代組裝,以及基因組對比軟件的檢出的準確性。SV檢測原理大致就是兩個基因組之間在某些片段是高度相似,比對上後針對於這些片段進行找對應位置是否有對應高度一致的序列。如果相對於參考基因組片段,比對的基因組沒有這段序列則為deletion變異;而相對於比對的基因組片段,參考基因沒有則為insersion變異。如果參考基因組與比對基因組在高度相似片段序列是處於翻轉的,則為inversion變異。
如果參考基因組與比對基因組在高度相似片段不是染色體位置對應附近,距離相隔比較遠,則為translocation變異。如果參考基因組與比對基因組上高度相似片段在其他位置也有,那為duplication變異。其中inversion、translocation變異還需要側翼序列一起來判定。
針對重測序,包括二代和三代的SV變異檢測與基因組差不多,除了使用軟件不一樣,最大不同是序列長度和只比對參考基因組上(非相互比對,僅單一比對到參考基因組),所以導致變異種類和數量會少一些,比如insertion可能檢測不到。
注:重測序SV檢測需要一定測序深度支持
分析內容
1.二代
二代重測序數據檢測SV主要用這兩個軟件lumpy-sv和delly軟件
Lumpy-sv軟件下載和安裝:GitHub - arq5x/lumpy-sv: lumpy: a general probabilistic framework for structural variant discovery
git clone --recursive https://github.com/arq5x/lumpy-sv.git cd lumpy-sv
make
cp bin/* /usr/local/bin/
delly軟件下載和安裝:GitHub - dellytools/delly: DELLY2: Structural variant discovery by integrated paired-end and split-read analysis
git clone --recursive https://github.com/dellytools/delly.git
cd delly/
make all
Delly軟件是專門SV預測方法,可以在short-reads中大規模基因分型和可視化的
串聯重複、倒位、易位、缺失等變異。它使用paired-ends、split-reads、read-depth來準確地描繪整個基因組中的基因組重排。結構變體可以使用delly-sansa進行註釋,並使用delly-maze或delly-suave進行可視化。
###
b=`cat rmdup.bam.list|tr "\n" ","`
b=${b%,}
s=`cat splitters.bam.list|tr "\n" ","`
s=${b%,}
d=`cat discordants.bam.list|tr "\n" ","`
d=${b%,}
lumpyexpress -B $b -S $s -D $d -o test_lumpy-sv.vcf
#
rmdup.bam.list文件格式

splitters.bam.list文件格式
discordants.bam.list文件格式
#extractSplitReads_BwaMem為lumpy軟件中一個程序
samtools view -h test.rmdup.bam | extractSplitReads_BwaMem -i stdin | samtools view -Sb - >test.splitters.bam
samtools view -b -F 1294 test.rmdup.bam >test.discordants.bam
合併多個方法檢測出來的SV,可以用SURVIVOR軟件(三代測序中也有描述)
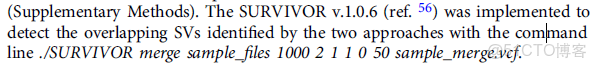
2.三代long-reads



3.基因組
2021cell水稻,2020cell大豆,2018science人
基因組檢測SV方法很多,主要是比對軟件差異,導致分析方法的差異。比對軟件有lastz,minimap2,mummer等,每種軟件原理不一,詳細去看軟件説明。主要講mummer比對,因為mummer比對最近比較流行的比對軟件,且基於比對的結果開發好幾種檢測SV的軟件,比如SVMU,SYRI等
3.1 mummer軟件
軟件下載:GitHub - mummer4/mummer: Mummer alignment tool
程序1:nucmer
nucmer是基於fasta數據文件之間進行比對的。它最適用於可能有大量重排的相似序列。比如:比較兩個基因組組裝,將組裝或測序reads mapping到已經完成的基因組,以及比較可能有大量重排和重複的相關物種的兩個基因組。
USAGE:
nucmer [options] <reference> <query>
[options] type 'nucmer -h' for a list of options.
<reference> specifies the multi-FastA sequence file that contains
the reference sequences, to be aligned with the queries.
<query> specifies the multi-FastA sequence file that contains
the query sequences, to be aligned with the references.
OUTPUT:
out.delta the delta encoded alignments between the reference and
query sequences. This file can be parsed with any of
the show-* programs which are described in the "RUNNING
THE MUMmer UTILITIES" section.
nucmer -c 1000 --mum --maxgap=1000 --prefix=ref_qry --threads=20 ref.fa qry.fa
程序2:dnadiff
dnadiff是基於nucmer的結果,輸出是的比對統計、snp、斷點。它主要是評估兩個高度結構相似序列的相似集。
USAGE: dnadiff [options] <reference> <query>
or dnadiff [options] -d <delta file>
<reference> Set the input reference multi-FASTA filename
<query> Set the input query multi-FASTA filename
or
<delta file> Unfiltered .delta alignment file from nucmer
OUTPUT:
.report - Summary of alignments, differences and SNPs
.delta - Standard nucmer alignment output
.1delta - 1-to-1 alignment from delta-filter -1
.mdelta - M-to-M alignment from delta-filter -m
.1coords - 1-to-1 coordinates from show-coords -THrcl .1delta
.mcoords - M-to-M coordinates from show-coords -THrcl .mdelta
.snps - SNPs from show-snps -rlTHC .1delta
.rdiff - Classified ref breakpoints from show-diff -rH .mdelta
.qdiff - Classified qry breakpoints from show-diff -qH .mdelta
.unref - Unaligned reference IDs and lengths (if applicable)
.unqry - Unaligned query IDs and lengths (if applicable)
dnadiff -d ref_qry.filted.delta -p ref_qry.filted.delta
程序3:delta-filter
該程序過濾由nucmer或promer生成的delta對齊文件,只留下所需要的對齊,這些對齊以與輸入相同的delta格式輸出到stdout。它的主要功能是LIS算法,它計算最長遞增的比對子集。這允許使用-g選項或局部一致的-1或-m計算一組全局對齊(即1對1和相互一致的順序)。可以使用-r將參考序列映射到查詢序列,或使用-q將查詢映射到參考。這允許用户排隊機會並重復誘導比對,只留下兩個數據集之間的“最佳”比對。
USAGE:
delta-filter [options] <deltafile>
[options] type 'delta-filter -h' for a list of options.
<deltafile> the .delta output file from either nucmer or promer.
OUTPUT:
stdout The same delta alignment format as output by nucmer and promer.
delta-filter -i 90 -l 1000 -1 ref_qry.delta > ref_qry.filted.delta
程序4:show-coords
該程序解析nucmer和promer的delta對齊輸出,並顯示座標以及有關對齊的其他有用信息。-c和-l選項在比較兩組組裝contig時很有用,因為這些選項有助於確定對齊是否跨越整個contig,或者只是對不同讀取的部分全中。當我們希望識別兩個基因組之間的共線區域,但對實際的比對相似性或外觀並不特別感興趣時,-b選項很有用。
USAGE:
show-coords [options] <deltafile>
[options] type 'show-coords -h' for a list of options.
<deltafile> the .delta output file from either nucmer or promer.
OUTPUT:
stdout run 'show-coords' without the -H option to see the column
header tags. Here is a description of each tag. Note that
some of the below tags do not apply to nucmer data, and that
all coordinates are inclusive and relative to the forward DNA
strand.
[S1] Start of the alignment region in the reference sequence.
[E1] End of the alignment region in the reference sequence.
[S2] Start of the alignment region in the query sequence.
[E2] End of the alignment region in the query sequence.
[LEN 1] Length of the alignment region in the reference sequence,
measured in nucleotides.
[LEN 2] Length of the alignment region in the query sequence, measured
in nucleotides.
[% IDY] Percent identity of the alignment, calculated as the
(number of exact matches) / ([LEN 1] + insertions in the query).
[% SIM] Percent similarity of the alignment, calculated like the above
value, but counting positive BLOSUM matrix scores instead of exact
matches.
[% STP] Percent of stop codons of the alignment, calculated as
(number of stop codons) / (([LEN 1] + insertions in the query) * 2).
[LEN R] Length of the reference sequence.
[LEN Q] Length of the query sequence.
[COV R] Percent coverage of the alignment on the reference sequence,
calculated as [LEN 1] / [LEN R].
[COV Q] Percent coverage of the alignment on the query sequence,
calculated as [LEN 2] / [LEN Q].
[FRM] Reading frame for the reference sequence and the reading frame
for the query sequence respectively. This is one of the columns
absent from the nucmer data, however, match direction can easily be
determined by the start and end coordinates.
[TAGS] The reference FastA ID and the query FastA ID.
There is also an optional final column (turned on with the -w
or -o option) that will contain some 'annotations'. The -o option will
annotate alignments that represent overlaps between two sequences,
while the -w option is antiquated and should no longer be used.
Sometimes, nucmer or promer will extend adjacent clusters past one
another, thus causing a somewhat redundant output, this option will
notify users of such rare occurrences.
show-coords -THrd ref_qry.filted.delta >ref_qry.filted.delta.coord
最終得到結果會有以下格式文件:

3.2 SVMU軟件
該軟件可以用lastz結果,也可以用mummer結果進行檢測PAV和CNV。SVMU全名是Structural Variants from MUmmer。
svmu sam2ref.mm.delta ref.fasta sample.fasta snp_mode sam_lastz.txt prefix
其中
snp_mode有兩種選擇 'h'或者'l'。h = report SNPs; l = no SNPs
prefix為輸出文件的前綴.
sv.prefix.txt為一個製表符分隔的文件,彙總了比對基因組相對於參考基因組的SV(PAV,CNV,INV)
small.prefix.txt為一個製表符分隔的文件,包含SNP和小片段的INDEL變異
cnv_all.prefix.txt為一個製表符分隔的文件,其中包含在比對基因組中以較高拷貝數(>1)存在的所有參考基因組區域。名稱中帶有“trans”代表它是轉座子或不同染色體中基因的非TE拷貝
cm.prefix.txt包含兩個基因組之間共線的參考基因組區域的文件
svmu ref_qry.filted.delta ref.fa qry.fa h null ref_qry
3.3 syri軟件
下載路徑:GitHub - schneebergerlab/syri: Synteny and Rearrangement Identifier
syri軟件主要是基於mummer的結果開發的一套檢測染色體級別變異的工具,變異包含SNP、INDEL、PAV、INV、TRANS等。
SyRI是從識別最長的共線性區域開始的,而對於所有非共線性區域都是通過對應在兩個基因組之間共線區域來判定重排,因此鑑定共線區域也會同時鑑定了所有結構重排。以此為基礎識別所有SR,將SR分類成各種變異,如倒位,易位或重複。此外,syri還可以識別所有共線性區域和重排區域內的局部變異。Local variations 包括小變異(如SNP和小indel),structural variations(如大indel,CNV(拷貝數變異)和HDR)。從比對結果中解析出短的變異,通過比較共線性區域或重排區域的連續排列之間的 overlaps 和 gaps 來預測結構變異。
syri軟件本身限制因素很多,主要是基因組必須是染色體版本、共線染色體需要都是正義鏈或都是負義鏈、染色體號需要一一對應等
syri軟件安裝也比較麻煩,需要是python3.5的環境,可以用conda安裝需要支持的包